
Un nouveau né de sexe féminin, né à terme, avec un poids de naissance de 3kg200 et un score d’Apgar calculé à 10 à 2 et 10 min. A l’examen, le bébé n’avait pas de dysmorphie faciale et les choanes étaient perméables.
Elle est issue d’une grossesse mal suivie, sans incident. Une seule échographie prénatale est réalisée déclarée normale.
Le nouveau né a développé, à H24 de vie une détresse respiratoire néonatale, avec des accès de cyanose.
A l’examen, elle était polypnéique avec une fréquence respiratoire à 80 cycles par minute, et un Score de Silverman calculé à 6. La saturation transcutanée d’oxygène (SaO2) était faible à l'air ambiant. Une cyanose généralisée était diagnostiquée. L’auscultation cardiaque était normale et il n’y avait pas de signe de défaillance cardiaque.
Les pouls fémoraux étaient très faibles.
Une échographie cardiaque transthoracique et un angioscanner thoracique ont été réalisés. Voici les clichés.
En examinant les clichés, vous retenez quel diagnostic ?
- Une tétralogie de Fallot
- Une coarctation de l’aorte
- Une Atrésie pulmonaire
- Une interruption de l’arche aortique
- Un retour veineux pulmonaire anormal
Réponse :
L'interruption de l’arche aortique (IAA) est une anomalie congénitale rare décrite pour la première fois par Abbott en 1927. Elle représente 3 pour un million de naissance vivante. Cette anomalie se caractérise par une absence totale de continuité anatomique entre l’arc aortique transversal (segment II de l’aorte) et l’arc thoracique descendant (segment III de l’aorte). En fonction du siège de cette interruption, Celoria et Patton ont classé cette anomalie en trois types A, B et C. Pour le type A, l’interruption est localisée au niveau de l’isthme de l’aorte, sous l’artère sous-clavière comme c’était le cas chez notre patient. Le type B est le plus fréquent. L’interruption est alors localisée entre la sous-clavière et la carotide gauche. Alors que pour le type C, cette interruption se situe entre la carotide gauche et le tronc artériel brachiocéphalique. L’IAA s’associe communément à d’autres malformations cardiaques, telle une communication inter ventriculaire ou une bicuspidie aortique. Dans notre cas, aucune de ces anomalies n’a été détectée. Dans la majorité des cas, ainsi que dans le notre, l’IAA s’accompagne d’un canal artériel perméable permettant une circulation de l’artère pulmonaire vers l’aorte descendante. C’est ce qui explique que durant la période néonatale, l’IAA est découverte dans le cadre de l’exploration d’un souffle cardiaque ou d’une cyanose. Notre patient présentait une cyanose généralisée. Mais le plus souvent, il s’agit d’un tableau de collapsus cardiovasculaire aigu ou de défaillance cardiaque après la fermeture spontanée du canal artériel. C’est pour cette raison que certaines équipes prescrivent les prostaglandines une fois l’IAA est diagnostiquée pour améliorer la tolérance de cette anomalie en attendant le traitement définitif.
La survie sans persistance du canal artériel a été parfois décrite. L’Imagerie en coupes représentée par l’échographie, l’angioscanner et l’IRM ont révolutionné le diagnostic de cette anomalie. Bien que l’échocardiographie offre une excellente visualisation de la majorité des anomalies de l’arc aortique, l’angioscanner ou l’IRM cardiaque demeurent nécessaires pour la confirmation du diagnostic, le bilan lésionnel complet en vue de la planification chirurgicale. Comme chez notre patient, le diagnostic d’IAA est retenu devant l’absence de continuité entre l’aorte descente et l'aorte ascendante. L’aorte descendante fait suite alors à l'artère pulmonaire par l'intermédiaire d’un canal artériel persistant dilaté.
La coarctation aortique représente le principal diagnostic différentiel. Elle est évoquée devant des pouls fémoraux faibles ou abolis. Mais l’angioscanner permet de redresser le diagnostic. La chirurgie cardiaque est le seul traitement. Le principe est l’élimination de la ducto-dépendance de la circulation systémique par une anastomose termino-terminale entre l’aorte descendante et l’arche aortique. Dans la plupart des cas, la récupération complète est possible. La mortalité chirurgicale rapportée dans la littérature est actuellement inférieure à 5 %. L’IAA peut être sporadique mais l’association avec le syndrome de Di George (délétion 22q11) est très fréquente. Dans le cas que nous présentons, le bébé n’avait pas de dysmorphie faciale et le caryotype était normal.
Conclusion
Bien que rare, l’IAA doit être inclues dans ces diagnostics différentiels d’une cyanose néonatale. L’échographie permet le diagnostic qui sera confirmé par l’angioscanner ou l’IRM. La chirurgie est le traitement de référence et une enquête génétique est indispensable à la recherche d’un syndrome de Di George.
Références consultées:
- Xiujie Tang, X., Lianyi Wang L., Wu Q., Tong X. Persistent Fifth Aortic Arch with Interrupted Aortic Arch. Card Surg 2015;30:284–7
- Celoria GC, Patton RB. Congenital absence of the aortic arch. Am. Heart J 1959;58(3):407-13
- Chen X., Qu YJ., Peng ZY., Lu JG., Ma X. Diagnosis of Congenital Aortic Arch Anomalies in Chinese Children by Multi-Detector Computed Tomography Angiography. J Huazhong Univ Sci Technol 2013;33(3):447-51
- Charakida M., Greil GF., Anderson D., Krasemann T. Unusual Diff erential Cyanosis in a Newborn due to an Interruption of the Aortic Arch. Klin Padiatr 2013; 225: 89–90
- Carissa M. Baker-Smith, MD, Angelo S. Milazzo, MD, Donald P. Frush, MD, James Jaggers, MD,
- Baker-Smith CL., Milazzo AS., Frush DP., Jaggers J, Kirby ML., Kanter RJ., Barker PC., et al. Double Aortic Arch with Aortic Atresia and Left-Sided Type B Interruption. Congenit Heart Dis. 2010;5:316–320
- Shirani S., Soleymanzadeh M. Diagnosis of aortic interruption by CT angiography. Pol J Radiol, 2013; 78(1): 72-74
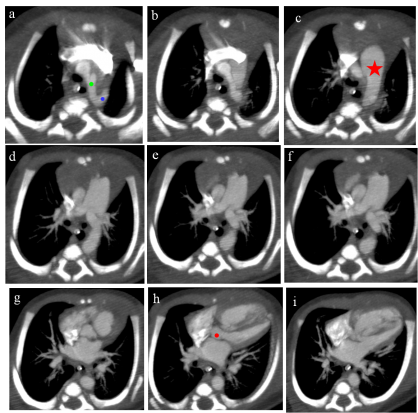
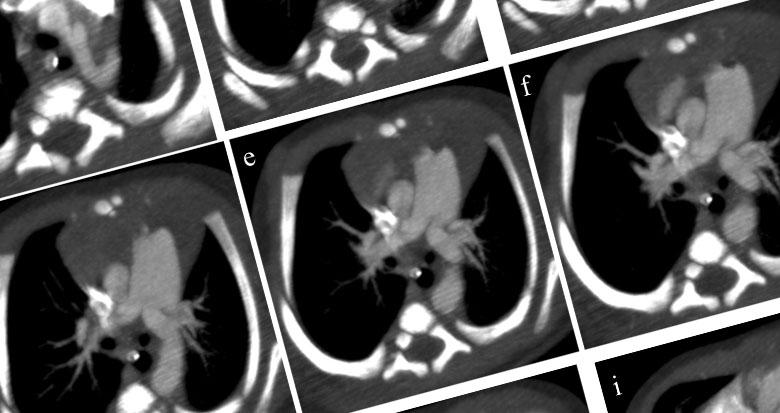
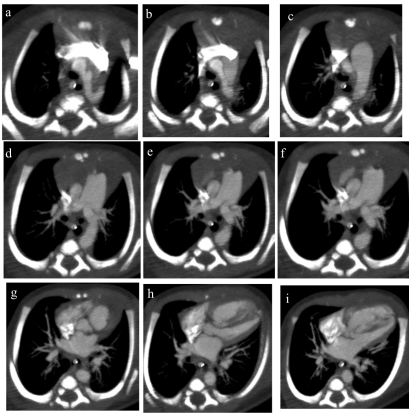
Figure 1 (de a jusqu’à i) : Reconstructions MIP (Maximum Intensity Projection) de coupes axiales successives d’un angioscanner thoracique.
L’aorte ascendante part normalement du ventricule gauche (point rouge). Le segment II de l’aorte donne trois troncs supra-aortiques (point vert), et il devient borgne après le départ de l’artère sous clavière gauche (point bleu). Absence de continuité entre l’aorte descente et l'aorte ascendante. L’aorte descendante fait suite à l'artère pulmonaire par l'intermédiaire d’un canal artériel persistant dilaté (grande étoile rouge).
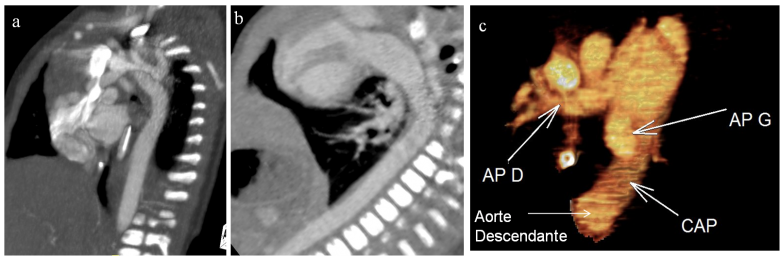
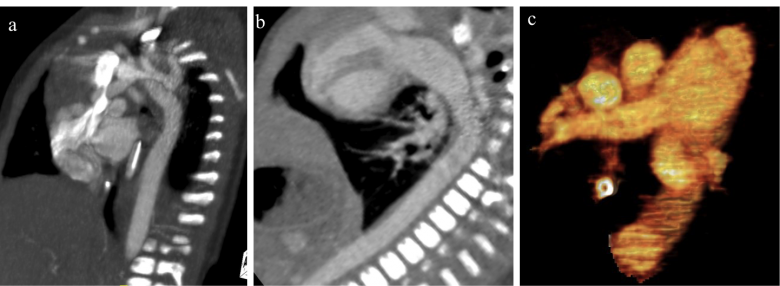
Figure 2 (a, b, c) : Reconstructions multiplanaires sagittales obliques (a et b) et Volume Rendering (c) confirmant les constatations des coupes axiales.
R. Salem, I. Melki, K. Ben Ameur, B. Hmida, S. Hamdi
Hôpital Fattouma Bourguiba, Monastir, Tunisie





